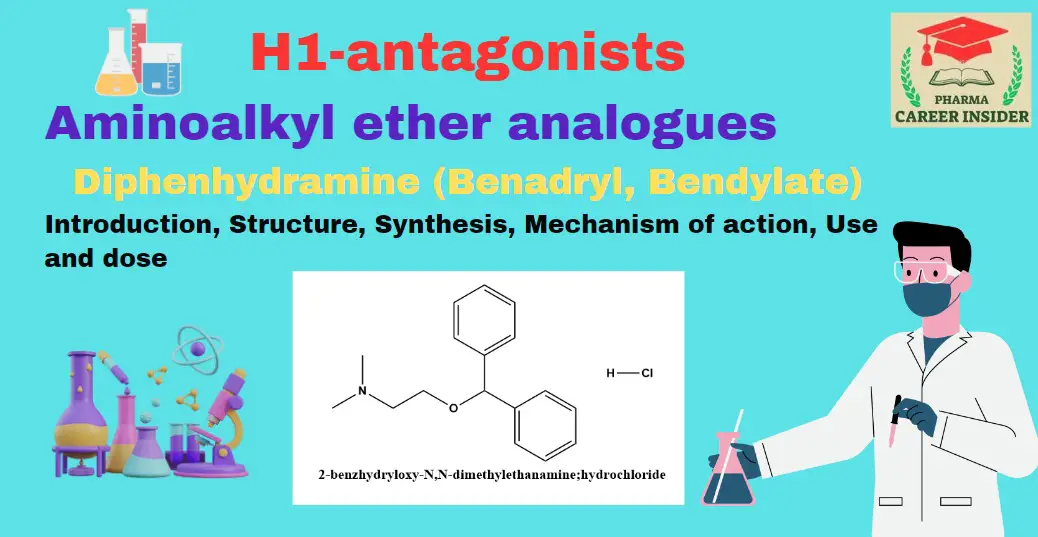
Introduction
Impurities in medicinal agents pose significant risks to drug safety, effectiveness, and overall stability. These contaminants can emerge from multiple phases of drug development and distribution, including the sourcing of raw materials, the intricacies of manufacturing processes, improper storage conditions, and inadequate packaging practices. During raw material sourcing, impurities may be introduced from substandard ingredients or contaminated supply chains, highlighting the importance of rigorous quality control measures. In the manufacturing process, factors such as equipment cleanliness, process validation, and environmental controls play crucial roles in minimizing contamination risks.
Additionally, improper storage conditions such as exposure to extreme temperatures, humidity, or light can lead to the degradation of the active ingredients and the formation of harmful byproducts. Furthermore, packaging materials themselves can be a source of impurities if they are not compatible with the medicinal agents they contain. Each of these stages requires careful attention and strict adherence to regulatory guidelines to safeguard the quality of pharmaceuticals. A comprehensive understanding of the origins and types of impurities is vital for pharmaceutical companies, regulatory bodies, and healthcare professionals.
This knowledge enables the development of strategies to prevent contamination and ensures the production of safe, effective, and high-quality medications for patients.
Types and Sources of Impurities
1. Impurities from Raw Materials
The quality of raw materials directly influences the purity of medicinal products. Impurities in raw materials may arise from:
- Contaminants in active pharmaceutical ingredients (APIs)
- Residual solvents used during synthesis
- Heavy metals present in natural sources
- Agricultural residues in herbal medicines
2. Process-Related Impurities
These impurities originate during manufacturing and include:
- By-products: Formed due to incomplete reactions or side reactions.
- Degradation Products: Resulting from thermal, oxidative, or hydrolytic decomposition of APIs.
- Reagents and catalysts: leftover chemicals from synthesis processes.
- Residual Solvents: Used in synthesis but not completely removed during purification.
3. Formulation-Related Impurities
Impurities may develop during the formulation of the final pharmaceutical product, such as:
- Excipient Degradation: Breakdown of excipients like preservatives, binders, or stabilizers.
- Drug-Excipient Interaction: Unintended chemical reactions between APIs and excipients.
- Leachable and extractable impurities: substances migrating from packaging materials into the drug product.
4. Storage and Stability-Related Impurities
Medicinal agents may degrade over time due to various environmental factors, leading to:
- Oxidation: Exposure to oxygen causing degradation.
- Hydrolysis: Interaction with moisture leading to breakdown.
- Photolysis: Light-induced degradation of drug molecules.
- Temperature-Related Degradation: High temperatures accelerate chemical breakdown.
5. Contaminants from Packaging Materials
Packaging plays a vital role in maintaining drug stability, but improper packaging can introduce impurities.
- Plastic and Glass Leachables: Migration of chemicals from packaging materials into the drug.
- Metal Contaminants: From improperly coated or corroded packaging components.
- Microbial contaminants: growth of bacteria or fungi due to inadequate storage conditions.
- conditions.
6. Environmental Contaminants
During production, storage, or transportation, pharmaceutical products can be exposed to:
- Airborne contaminants: dust, microorganisms, and particulate matter.
- Waterborne Impurities: Contaminated water used in manufacturing processes.
- Cross-contamination: accidental mixing of different drug compounds in multi-product facilities.

Regulatory Guidelines for Controlling Impurities
To ensure the safety and efficacy of medicinal products, regulatory authorities such as the U.S. Food and Drug Administration (FDA), European Medicines Agency (EMA), and International Council for Harmonization (ICH) have established guidelines for impurity assessment and control. Key regulatory frameworks include:
- ICH Q3A: Guidelines for impurities in new drug substances.
- ICH Q3B: Impurity guidelines for new drug products.
- ICH Q3C: Guidelines for residual solvents in pharmaceuticals.
- Good Manufacturing Practices (GMP): Ensuring controlled production environments.
Strategies for Impurity Control and Reduction
Pharmaceutical industries must implement strict quality control measures to minimize impurities. Effective strategies include:
- Raw Material Selection: Using high-quality APIs and excipients with minimal impurities.
- Optimized Manufacturing Processes: Employing advanced synthesis methods to reduce by-products.
- Analytical Testing: Utilizing chromatographic and spectroscopic techniques for impurity detection.
- Proper Storage Conditions: Maintaining optimal temperature, humidity, and light exposure conditions.
- Robust Packaging: Using inert and stable packaging materials to prevent contamination.
- Compliance with Regulatory Standards: Adhering to international guidelines to ensure drug purity and safety.
Conclusion
The presence of impurities in medicinal agents poses significant risks to drug safety and efficacy. Identifying, controlling, and minimizing impurities is critical for pharmaceutical industries to meet regulatory standards and ensure patient safety. With stringent quality control, optimized manufacturing processes, and adherence to international guidelines, the pharmaceutical industry can produce high-purity and effective medicinal products.
By understanding the sources of impurities in medicinal agents, researchers and manufacturers can take proactive measures to mitigate contamination risks, ensuring high-quality pharmaceutical products for global healthcare.